MMsINC Structure Search allows user to query the databse compounds using a chemical structure (drawn by JME)
or using the structural query input (SMILES, MMscode, InChI, Molecular Formula).
You can search structure using different type of query: identical structure, substructure or similarity search.
Identical Structure search retrieves the compound that contains the exact chemical structure. In some case, this request displays also all the isomers of the structure, which is why you can see more than one result.
Substructure search retrieves compounds that contain a chemical structural pattern defined by you, called substructure. Each molecule retrieved must contain the substructure.
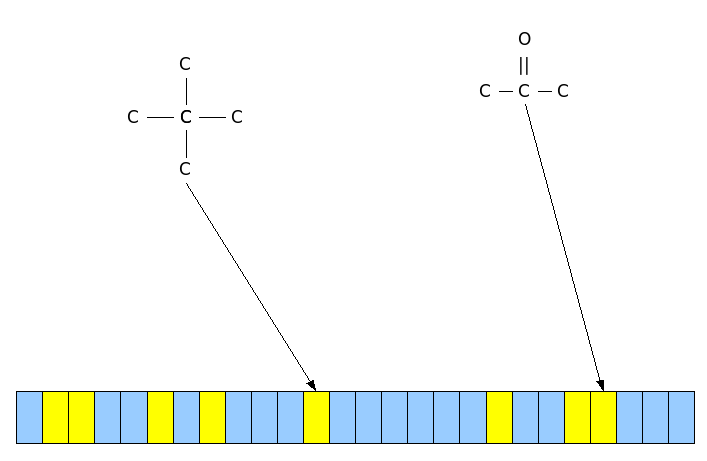
Scissoring search retrieves molecules that contain fragments common with those extracted by the query. Users can choose which fragments will be used and the type of research (AND/OR).
Similarity search retrieves similar compounds to the chemical structure given in input, using the tanimoto co-efficient. The similarity is measured using the Tanimoto equation and a binary fingerprint, called molecular fingerprint, computed for every structure. A molecular fingerprint is a bit sequence where
each bit in the fingerprint (or fragment bit-string) represents one molecular fragment.
Molecular fingerprint. The yellow represents the presence of the fragment in the molecule whereas the blue the abscence.
The bit string for a molecule records the presence (1) or absence (0) of each fragment in the molecule. The fingerprint-based similarity is the comparison between two fingerprints to find the common bits, and then compute the Tanimoto coefficient.
The number of similar compounds extracted depends on the threshold value of the tanimoto co-efficient. The values available for the tanimoto are 0.85, 0.90 and 0.95.
SMILES: The "simplified molecular input line entry specification" or SMILES is a specification to describe unambiguously the structure of chemical molecules using short ASCII strings.
MMscode: MMsINC Compound Identification, a non-zero integer MMsINC accession identifier for a unique chemical structure.
InChI: The "IUPAC International Chemical Identifier" or InChI is a textual identifier for chemical substances, designed to provide a standard and human-readable way to encode molecular information and to facilitate the search for such information in databases and on the web.
Molecular Formula: A chemical formula is a concise way of expressing information about the atoms that constitute a particular chemical compound.
For molecular compounds, it identifies each constituent element by its chemical symbol and indicates the number of atoms of each element found in each discrete molecule of that compound.
Molecular weight (MW): (including implicit hydrogens) in atomic mass units with atomic weights taken from [/CRC Handbook of Chemistry and Physics/. CRC Press (1994)]. The unit of each molecular weight is g/mol.
logS: Log of the aqueous solubility (mol/L).This property is calculated from an atom contribution linear atom type model [Hou 2004] with r2 = 0.90, ~1,200 molecules.
SlogP: Log of the octanol/water partition coefficient (including implicit hydrogens). This property is an atomic contribution model [Crippen 1999] that calculates logP from the given structure; i.e., the correct protonation state (washed structures). Results may vary from the logP(o/w) descriptor. The training set for SlogP was ~7000 structures./li>
Reactive groups: Indicator of the presence of reactive groups. A non-zero value indicates that the molecule contains a reactive group. The table of reactive groups is based on the Oprea set [Oprea /J. Comp. Aid. Mol. Des./ *14*, 251-264 (2000)] and includes metals, phospho-, N/O/S-N/O/S single bonds, thiols, acyl halides, Michael Acceptors, azides, esters, etc.
Topological Properties
Globularity, or inverse condition number (smallest eigenvalue divided by the largest eigenvalue) of the covariance matrix of atomic coordinates. A value of 1 indicates a perfect sphere while a value of 0 indicates a two- or one-dimensional object.
Sterimol/B1-4: steric distances perpendicular to the bond axis. These define a bounding box for the substituent and are numbered in ascending size order.
Sterimol/L: steric length parameter, measured along the substitution point bond axis.
Surface and Volume Properties
ASA: water accessible surface area calculated using a radius of 1.4 A for the water molecule. A polyhedral representation is used for each atom in calculating the surface area.
ASA+: water accessible surface area of all atoms with positive partial charge (strictly greater than 0).
ASA-: water accessible surface area of all atoms with negative partial charge (strictly less than 0).
ASA_H: Water accessible surface area of all hydrophobic (|/q_i /|<0.2) atoms.
Volume: van der Waals volume calculated using a grid approximation (spacing 0.75 A).
Pharmacophoric Properties
HB donor groups: Number of hydrogen bond donor atoms (not counting basic atoms but counting atoms that are both hydrogen bond donors and acceptors such as -OH).
HB acceptor groups: Number of hydrogen bond acceptor atoms (not counting acidic atoms but counting atoms that are both hydrogen bond donors and acceptors such as -OH).
Acid groups: Number of acidic atoms.
Basic groups: Number of basic atoms.
Chiral centers: Number of chiral centers.
Energetic Properties
Potential Energy (e): total potential energy, calculated from the 3D structure of the molecule, expressed in kcal/mol.
You could retrieve supplementary informations on the molecular descriptions, if you click Here.